En el blog anterior vimos que los tratamientos convencionales para enfermedades mentales están enfocados en la estabilización de los neurotransmisores, sin embargo, hasta un 30% de las depresiones son resistentes al tratamiento, lo que obliga a pensar que otras vías fisiopatológicas están involucradas en este tipo de enfermedades.
El modelo actual entiende estos trastornos como enfermedades sistémicas, donde el cerebro es altamente sensible a alteraciones metabólicas, inmunes y mitocondriales.
Se ha postulado que las enfermedades mentales también pueden ocurrir por los siguientes factores:
- Resistencia a la insulina.
- Inflamación crónica de bajo grado.
- Estrés oxidativo.
- Vía de la Quinurenina.
- Alteraciones mitocondriales.
En el blog de hoy profundizaremos el concepto de resistencia a la insulina cerebral y su relación con las enfermedades mentales.
Resistencia a la Insulina.
Las neuronas son en gran medida intolerantes a un suministro energético inadecuado, por lo que la alta demanda energética del cerebro lo predispone a diversas enfermedades si se interrumpe dicho suministro.
Una gran variedad de patologías del sistema nervioso central son consecuencia, y en ocasiones también causa, de alteraciones del metabolismo energético de la glucosa a nivel central o periférico, que pueden verse afectadas en casi todos los niveles de las cascadas metabólicas celulares o bioquímicas.
Tanto la diabetes tipo 1 como la tipo 2 se asocian con un mayor riesgo de deterioro cognitivo relacionado con la edad y trastornos del estado de ánimo, especialmente depresión. Se ha demostrado que la acción de la insulina regula la señalización y la plasticidad neuronal.
No obstante, no hace mucho tiempo se ha estado hablando de la resistencia a la Insulina cerebral también llamada Diabetes tipo 3, con la cual se ha establecido una correlación más directa en cuanto a su influencia en la aparición de enfermedades mentales.
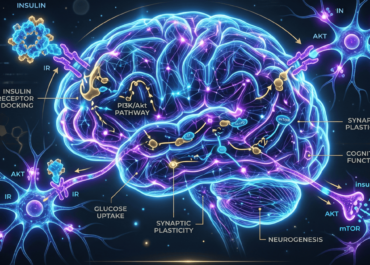
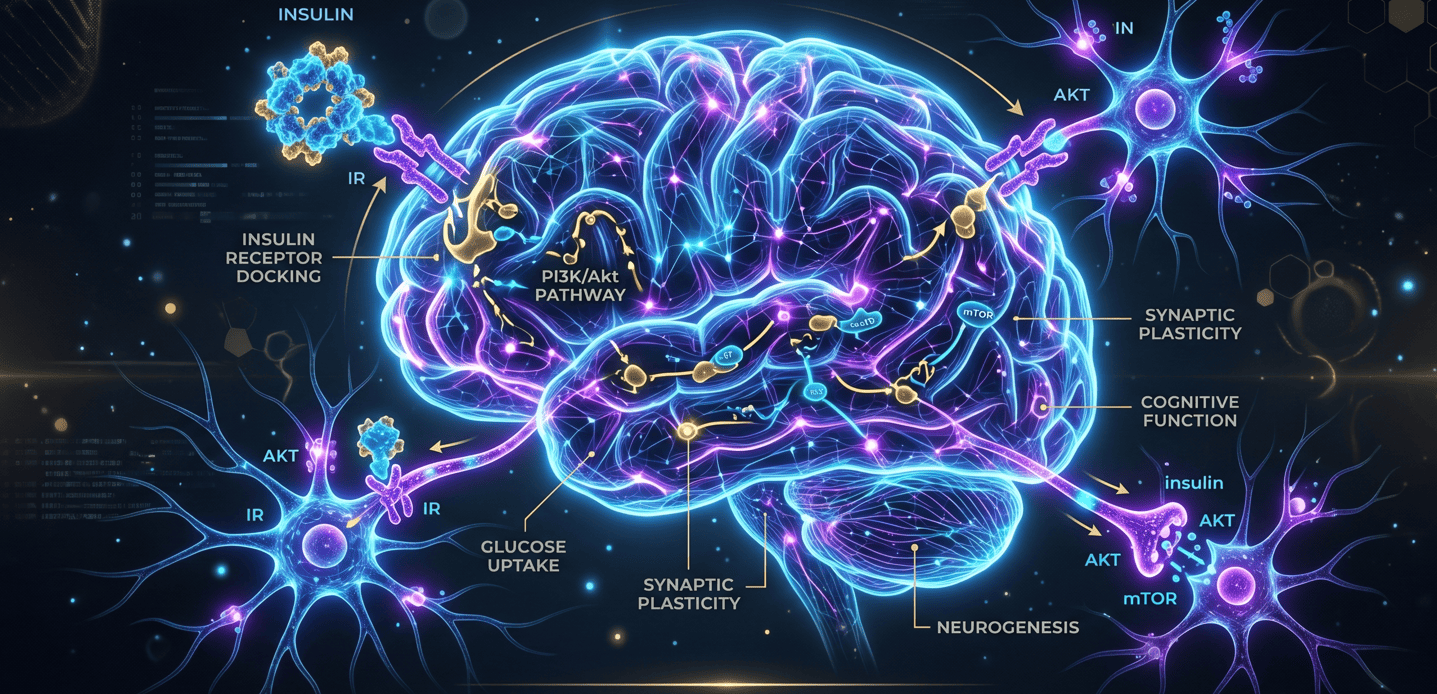
Imagen 1. Infografía de la Resistencia a la Insulina cerebral. Fuente: Generado por IA.

Tabla 1. Diferencia entre resistencia a la insulina (RI) periférica y central. Fuente: elaboración propia.
Diferencias | RI Periférica (Diabetes tipo 2) | RI Cerebral (Diabetes tipo 3) |
Órgano o tejido afectado | Músculos, hígado, tejido adiposo Eleva glucosa en sangres | Neuronas y células gliales del SNC |
Mecanismo | Las células no captan glucosa eficientemente, el páncreas produce más insulina para compensar | Señalización de insulina interrumpida en el cerebro. Reduce la captación de glucosa neuronal y la plasticidad sináptica |
Consecuencias | Hiperglucemia Obesidad Diabetes tipo 2 Hígado Graso | Deterioro cognitivo Pérdida de memoria, “niebla mental” Neuroinflamación Mayor riesgo de enfermedad de Alzheimer |
La señalización de insulina interrumpida en el cerebro (o resistencia a la insulina cerebral) significa que las neuronas no responden adecuadamente a esta hormona, afectando el metabolismo energético, la función cognitiva y la plasticidad neuronal.
La resistencia cerebral puede aparecer mucho antes que la periférica, incluso tras una sobrealimentación rápida, protegiendo inicialmente la periferia a costa de la función cognitiva.
Ambas formas están íntimamente ligadas por la obesidad, el estrés oxidativo y la inflamación crónica.
La resistencia a la insulina periférica puede, con el tiempo, aumentar el riesgo de desarrollar resistencia a la insulina en el cerebro al alterar la barrera hematoencefálica.
¿El cerebro es un órgano insulinodependiente?
A diferencia de los músculos o el tejido adiposo, la gran mayoría de las neuronas no necesitan insulina para que la glucosa entre en ellas.
Esto es una ventaja evolutiva crítica: el cerebro consume aproximadamente el 20% de la energía total del cuerpo y no puede permitirse esperar a que los niveles de insulina suban para obtener «combustible».
En los humanos, el cerebro representa el 2% del peso corporal, pero consume el 20% de la energía derivada de la glucosa, lo que lo convierte en el principal consumidor de glucosa (5,6 mg de glucosa por 100 g de tejido cerebral humano por minuto).
Sin embargo, hay otras áreas del cerebro que, si son muy sensibles a la insulina como el hipocampo, la corteza prefrontal y el sistema límbico.
Captación de glucosa en el cerebro.
Imagen 2 Esquemas representativos del cerebro, dendritas y axones y barrera hematoencefálica (BHE).

Para la captación de la glucosa por el cerebro son fundamentales los siguientes elementos:
- La Barrera hematoencefálica.
- Mecanismo de difusión facilitada.
- Los Transportadores específicos de glucosa.
La glucosa entra al cerebro principalmente a través de la barrera hematoencefálica (BHE) mediante un proceso de difusión facilitada usando los Transportadores de glucosa específicos, fundamentalmente GLUT1, el cual se encuentra en las células endoteliales de los capilares cerebrales, permitiendo que la glucosa pase de la sangre al tejido cerebral para nutrir neuronas y astrocitos.
La concentración de glucosa en el tejido cerebral en estado estacionario es de aproximadamente el 20% de la del plasma arterial.
El GLUT1 media además la captación de glucosa desde el líquido extracelular hacia los astrocitos, la oligodendroglía y la microglía.
Se ha evidenciado que más del 10% de la epilepsia de ausencia de inicio temprano y hasta el 1% de las epilepsias generalizadas idiopáticas comunes se han atribuido a la deficiencia de GLUT1 causadas por mutaciones en el gen SLC2A1.
Una vez en el cerebro, la glucosa es captada por las neuronas, principalmente a través de transportadores GLUT3, este transportador se encuentra altamente concentrado en axones, dendritas y en la membrana plasmática de los cuerpos celulares.
El GLUT3 es un transportador de alta afinidad independiente de insulina, crítico para el desarrollo y mantenimiento neuronal, mostrando abundancia 6-10 veces mayor que el GLUT1 en ciertas neuronas.
La expresión neuronal del GLUT3 aumenta en hipoxia-isquemia y está implicado en enfermedades como el Alzheimer, Huntington y epilepsia.
En el hipotálamo, el transportador GLUT2, actúa como sensor para detectar los niveles de glucosa en sangre y regular la ingesta de alimentos.
El hipocampo y el cerebelo, que si son regulados por insulina requieren los transportadores GLUT4/8.
Rol de los Astrocitos: Los astrocitos también captan glucosa para sus funciones y pueden almacenar glucógeno, o convertir la glucosa en lactato para suministrar energía a las neuronas.
Estos mecanismos aseguran que el cerebro reciba un flujo continuo de combustible crucial, ya que depende casi exclusivamente de la glucosa para funcionar.
Imagen 3. Representación de los astrocitos.

¿Qué ocurre cuando hay resistencia cerebral a la insulina?
- ↓ señalización PI3K-Akt (crucial en el crecimiento y supervivencia celular)
- ↓ BDNF
- ↓ plasticidad sináptica
- ↑ vulnerabilidad al estrés
- ↑ inflamación cerebral
- Deterioro en la memoria en pacientes con esquizofrenia
- Empeoramiento del estado cognitivo en pacientes con esquizofrenia
Relación de la Resistencia a la Insulina (RI) cerebral con enfermedades mentales
Tabla 2. Relación de Resistencia a la Insulina cerebral con enfermedades mentales. Fuente: elaboración propia.
Patología | Evidencia |
Depresión | 40-50% RI subclínica |
Ansiedad | Hipoglucemias reactivas Hiperactivación amigdalar |
Esquizofrenia | RI cerebral independiente de antipsicóticos Predisposición genética a niveles anormalmente altos de insulina en ayunas. Estudios post mortem–>expresión disminuida de los receptores de insulina
|
Autismo | Alteraciones tempranas de señalización insulina-IGF |
Trastorno Afectivo Bipolar | RI correlaciona con ciclos rápidos y resistencia |
La comorbilidad de la esquizofrenia y la diabetes tipo 2 (DM2) está bien establecida, siendo la DM2 y sus precursores (es decir, la resistencia a la insulina) más prevalentes en pacientes con esquizofrenia independientemente del tratamiento con antipsicóticos.
La prediabetes y la DM2 pueden desarrollarse como efectos secundarios de ciertos agentes antipsicóticos, particularmente antipsicóticos de segunda generación (es decir, olanzapina, clozapina, quetiapina, risperidona, aripiprazol, paliperidona, ziprasidona y lurasidona), que se prescriben comúnmente para el tratamiento de trastornos psicóticos como la esquizofrenia y el trastorno bipolar.
No obstante, lo anterior, pacientes sin tratamiento previo que experimentan su primer episodio de psicosis también han mostrado una mayor incidencia de prediabetes y DM2 en comparación con una población general de la misma edad.
Esta observación sugiere que el mayor riesgo de DM2 en la esquizofrenia puede no estar relacionado con los efectos secundarios inducidos por el uso de fármacos antipsicóticos.
Aspectos clave de esta condición: (de la señalización de insulina interrumpida).
- Mecanismo: El cerebro pierde la capacidad de usar la insulina para captar glucosa eficientemente, lo que altera la energía neuronal y aumenta el estrés oxidativo.
- Consecuencias cognitivas: Provoca deterioro de la memoria, problemas en la toma de decisiones y pérdida de plasticidad sináptica (conexiones entre neuronas).
- Vínculo con Alzheimer: Esta interrupción aumenta la acumulación de placa beta-amiloide y proteína tau, características principales del Alzheimer.
- Causas: Dieta poco saludable (alta en grasas/azúcares), sedentarismo, neuroinflamación y carga alostática (estrés crónico).
- Soluciones: El ejercicio físico (aeróbico/fuerza) y estilos de vida saludables pueden mejorar la sensibilidad a la insulina en el cerebro.
Como vimos en este blog, la resistencia a la insulina cerebral es un mecanismo fisiopatológico plausible para la expresión de las enfermedades mentales.
Doctora Marleny Beltrán Floriano.
Médica y Cirujana.
Diplomado en Medicina Estética.

Bibliografía.
- Calabrese V, Lodi R, Tonon C, D′Agata V, Sapienza M, Scapagnini G, et al. Estrés oxidativo, disfunción mitocondrial y respuesta al estrés celular en la ataxia de Friedreich. J Neurol Sci. 2005;233:145–162. doi: 10.1016/j.jns.2005.03.012. [ DOI ] [ PubMed ] [ Google Académico ]
- Munnich A, Rustin P. Espectro clínico y diagnóstico de los trastornos mitocondriales. Am J Med Genet. 2001;106:4–17. doi: 10.1002/ajmg.1391. [ DOI ] [ PubMed ] [ Google Académico ]
- Roberts RA, Laskin DL, Smith CV, Robertson FM, Allen EM, Doorn JA, et al. Estrés nitrativo y oxidativo en toxicología y enfermedad. Toxicol Sci. 2009;112:4–16. doi: 10.1093/toxsci/kfp179. [ DOI ] [ Artículo PMC gratuito ] [ PubMed ] [ Google Académico ]
- Ames A., III Metabolismo energético del SNC en relación con la función. Brain Res Brain Res Rev. 2000;34:42–68. doi: 10.1016/s0165-0173(00)00038-2. [ DOI ] [ PubMed ] [ Google Académico ]
- Mattson MP, Liu D. Energética y estrés oxidativo en la plasticidad sináptica y los trastornos neurodegenerativos. Neuromolecular Med. 2002;2:215–231. doi: 10.1385/NMM:2:2:215. [ DOI ] [ PubMed ] [ Google Académico ]
- Chen H, Chan DC. Dinámica mitocondrial: fusión, fisión, movimiento y mitofagia en enfermedades neurodegenerativas. Hum Mol Genet. 2009;18 (R2:R169–R176. doi: 10.1093/hmg/ddp326. [ DOI ] [ Artículo PMC gratuito ] [ PubMed ] [ Google Académico ]
- Li Z, Okamoto K, Hayashi Y, Sheng M. La importancia de las mitocondrias dendríticas en la morfogénesis y plasticidad de las espinas y sinapsis. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003.
- https://pmc.ncbi.nlm.nih.gov/articles/PMC3285768/
!Déjanos tu comentario!